
Background
5-hydroxytryptamine (5-HT; serotonin) is a neurotransmitter. 5-HT is critical for essentially all human behaviours, and is particularly important in controlling mood and perception.1 Given this, it is not surprising that compounds which target the same structures in the brain, such as LSD, are often psychedelic hallucinogens.1
Cells that respond to 5-HT are stimulated when the neurotransmitter binds to 5-HT receptors (5-HTRs) embedded in the cell membrane.1 The potential role of 5-HT signaling in antidepressant efficacy (though it is clearly not the whole story2) combined with early positive results from clinical trials of serotonergic psychedelics has spurred interest in targeting 5-HT2A receptors to develop new drugs for depression and other forms of mental illness.1,3 However, these drug discovery efforts have faced two key difficulties: firstly, new compounds must avoid activating the closely related 5-HT2B receptor, or risk potentially lethal cardiovascular adverse effects; new compounds must be sufficiently selective for 5-HT2A.4 Secondly, potential drugs would ideally activate 5-HT2A without producing the hallucinogenic effects of compounds such as LSD and psilocin, which until recently were considered not acceptable for antidepressants.5
In a recently published article, a large group of authors from several U.S. universities overcame both problems with a combination of extensive virtual screening – a computer-intensive method for simulating early drug discovery on a massive scale – and information from structural biology to produce truly novel compounds that could one day become medications.6
Generating a library of new compounds
Compounds which are known to activate the 5-HT2A receptor (agonists) fall into one of a few categories of chemical structures, some of which are naturally serotonin-like; unfortunately, existing compounds tend to be poorly selective for 5-HT2A, hallucinogenic, or both.7 LSD, a well-known agonist for 5-HT2A, contains a tetrahydropyridine (THP) chemical “motif” (Figure 1A). This motif is therefore, on the face of it, a good starting point from which to build a new 5-HT2A agonist, and the authors considered it underexplored. However, the mechanisms by which drugs bind to their targets are extraordinarily complex, and it is not yet possible to reliably design effective drugs from chemical structures alone.
Virtual screening is a powerful tool for overcoming this hurdle, whereby computer algorithms are used to generate many simple variations of compounds containing the THP backbone and then tested for suitability by various computational methods (Figure 1B). The authors employed this approach, filtering the results of the algorithm to those with conservative physical properties that would allow it to cross the blood-brain barrier (such as charge and size), producing a library of around 75 million candidate compounds with a THP motif.

Figure 1: A) The chemical structure of LSD, with the THP moiety highlighted in blue. B) Schematic of the workflow used by the authors to generate a large virtual library of THP-containing compounds and test them, first by computer simulations and later by biological and biochemical experiments. Adapted from Kaplan et al., 2022 by Kieran Deane-Alder.6
Computerized testing of compound suitability
It would be impossible to test all 75 million compounds for pharmacological effectiveness at 5-HT2A, so the authors used further virtual screening by computer. First, they tested for simple binding to 5-HT2A using a simulation of the receptor’s 3D structure by digitally mutating the closely-related 5-HT2B to match the amino acid sequence of 5-HT2A, to act as “bait” for the compounds. A staggering 7.46 trillion compound/receptor complexes were simulated, and narrowed down to a final set of 205 compounds after filtering for redundancy and similarity to existing compounds in the scientific literature.
From these 205 compounds, the authors selected a final set of 17 to actually synthesize and test in real-world experiments based on the variety of their chemical structures, to maximize the chance of finding a good ‘hit’. Of the 17 compounds, only four had any ability to bind to the 5-HT2A receptor (Figure 2A), though none activated it and compounds 26 and 27 were able to activate the related and undesirable 5-HT2B receptor.

Figure 2: A) The four (of 17) synthesized THP-containing compounds which were found to bind to 5-HT2 receptors. Different functional groups appended to the THP core (black) are coloured light green and pale blue. LSD (left) is provided for comparison. Compounds which were found to bind to 5-HT2A are in the grey box. B) Computer-predicted mechanisms of binding for each compound, with LSD for comparison. Ki: inhibition binding constant of the compound for a given receptor (a measure of affinity). EC50: the concentration of compound required to produce a half-maximal response (a measure of potency). Adapted from Kaplan et al., 2022 by Kieran Deane-Alder.6
By analyzing the predictions that their virtual screen made about how each of these four compounds binds to 5-HT2A (Figure 2B), the authors were able to rationally design new compounds that would activate the receptor selectively. The observation that the THP core component had the same folded 3D shape across the compounds was particularly valuable (Figure 2B). Using this information with an iterative medicinal chemistry approach, they produced three optimized compounds with specific enantiomers (“handedness” in 3D space) to adopt specific 3D shapes similar to those in the computer simulations: (S)-65, (R)-69 and (R)-70 (Figure 3A). (R)-69 and (R)-70 were found to be partial agonists of 5-HT2A, and computer simulations predicted similar binding interactions to the earlier compound series (Figure 3B).
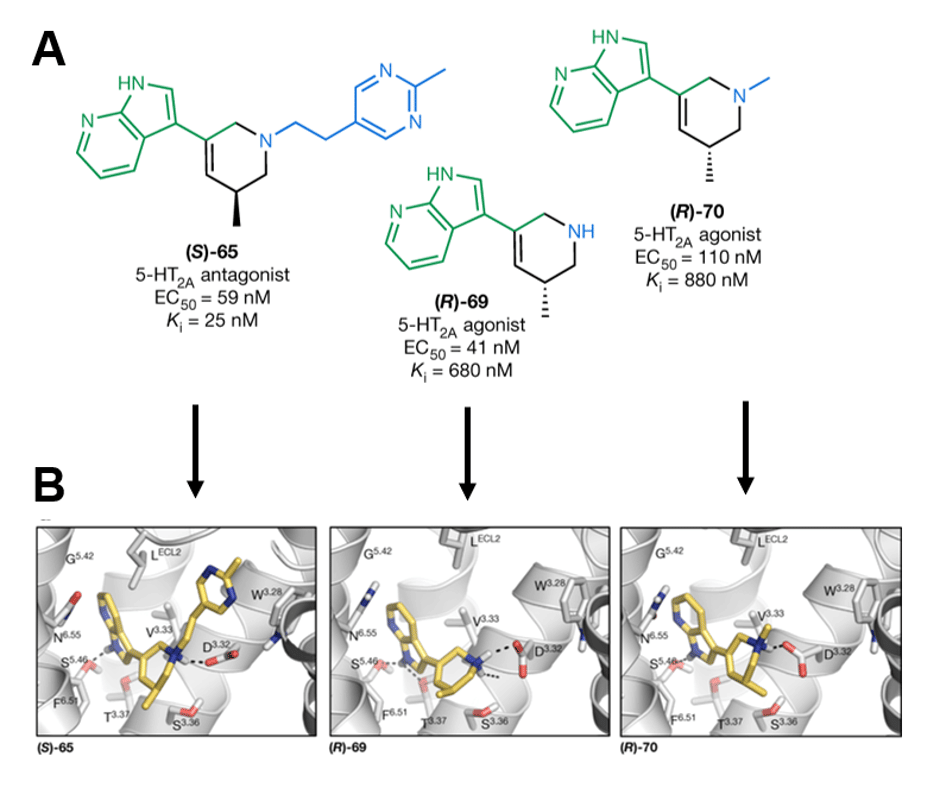
Figure 3: A) The three enantiospecific compounds developed from the earlier set of four synthesized compounds. B) Simulated binding arrangements for each compound. Adapted from Kaplan et al., 2022 by Kieran Deane-Alder (7).
Structural biology of 5-HT2A informs future development
To test the accuracy of their computer simulations, the authors also obtained the first cryogenic transmission electron microscopy (cryo-EM) structure of 5-HT2A bound to (R)-69 (Figure 4). Cryo-EM is the best technique for studying GPCR structure, because it allows researchers to observe the 3D shapes of protein complexes in an environment close to their native state. Remarkably, the predicted and experimentally observed binding of (R)-69 were almost identical. Compared to structures of 5-HT2B bound to compounds such as LSD, the authors also noted a unique pattern of binding for (R)-69.
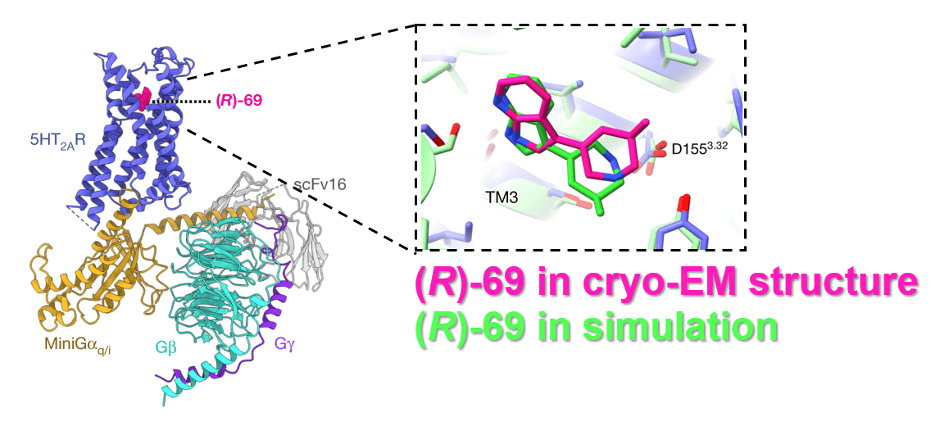
Figure 4: Left: Secondary structure cartoon of the 5-HT2A atomic model obtained by cryo-EM. The different components of the receptor (blue) and G protein complex are highlighted in different colours. Right: Cutaway of the primary binding site of the receptor, showing the position and orientation of compound (R)-69 in the cryo-EM structure (magenta) overlaid with that predicted by computer simulation (green). Adapted from Kaplan et al., 2022 by Kieran Deane-Alder.6
Animal testing of the new compounds
Finally, the authors used mouse models to investigate the behavioural effects of (R)-69 and (R)-70, which were found to easily cross the blood-brain barrier into the central nervous system. The compounds were assessed for the head-twitch response, a well-validated method for predicting psychedelic effects of substances. Compared to LSD, even at high doses the two new compounds induced little head twitch. Additionally, dosing mice with (R)-69 or (R)-70 after pre-treatment with LSD reduced head twitch, indicating they displaced LSD from 5-HT2A receptors.
In a subsequent experiment, mice that had been trained to show learned helplessness in response to mild electric shocks – an animal model of depressive symptoms – significantly improved on surrogates for mood (desire for sugar) and motivation (active avoidance of tail manipulation) after treatment with (R)-70. The magnitude of improvement for the new compounds was similar to psilocin, the active metabolite of psilocybin.
Conclusion
Overall, this paper demonstrates the most comprehensive case study yet for the power of high-throughput virtual screening combined with structure-informed drug design to overcome challenges posed by a difficult drug target. The authors have convincingly explored previously untapped chemical space to produce new 5-HT2A agonists, the first steps towards effective drugs at this receptor.

Kieran Deane-Alder
Kieran is a PhD candidate at the Monash Institute of Pharmaceutical Sciences in Victoria, Australia. His research employs structural biology methods to shed light on the diverse pharmacology of compounds that target the cannabinoid receptors. Kieran is deeply interested in all aspects of psychoactive compounds, from fundamental biochemistry to clinical safety.
What’s the paper and who are the authors?
I have Chroic Clusterheadache. As far as I understand molecular structure, the DMT molecule is strikingly similar to Sumatriptan.
Are the two related in any way? I read they bind to 5-Ht receptors? Does my neurologist give me a derivative of dmt? Sumatriptans flood the brain with seratonine and the pain goes away. Just wondering what these meds are doing to me…